The medical device approval process has become more difficult as FDA guidelines and compliance mandates grow increasingly stringent. As a result, many companies face the challenge of maintaining healthy growth and investment in research and development.
In a recent webinar organized by Cognizant Technology Solutions, three medical device industry experts – Ronnie Toddywala, Ph.D., MBA, CEO of Nostrum Technologies, LLC; Janet Trunzo, Senior Executive Vice President, Technology & Regulatory Affairs, AdvaMed; and Sriraman Nagarajan, Vice President,Life Sciences Practice, Cognizant, discussed trends and challenges facing this sector, and which ones will shape the device industry in the near future. We present some excerpts.

Regulatory updates and its impact
Janet Trunzo, AdvaMed: The medical device industry has continued to innovate in 2014, and is continuing to do so. There is a new focus on post-market data collection. FDA is continuing to issue key guidance documents intended to improve clinical trials and IDE to allow for expedited access to breakthrough technology. The FDA Safety and Innovation Act of 2012 not only reauthorized user fees for another five years, but also address several regulatory procedures. Along with modifications to the IDE and the 510(k) process, the Unique Device Identification requirement was finalized in the fall of 2013. UDI rule set forth a plan in which all devices will have a unique identification number within a specific timeframe, based on the risk classification. So far UDI rule has seen several implementation challenges – not only for the industry, but also for the agency. FDA has established its new UDI database. The agency has averaged over 500 implementation questions each month, and had often faced a delay in getting answers out for some of the more challenging questions.
Major industry challenges
Sriraman Nagarajan, Cognizant: Post the Affordable Care Act, there has been a major shift in focus on quality and reallocation of funds. There is an effort to trim down recurring spend on compliance And this is happening not just with across the portfolio of products, but also across technology platforms. There is a change in how we are addressing the market, shifting to value-based, evidence-based selling and this is creating a continuing downward pressure on price points. Payers and providers are continuing to evaluate devices on their performance, but on top of it are also looking at economic value of the products. Health reps are increasingly expected to have a better, and holistic view of their entire product portfolio, and a lot of our customers are preparing for these changes.
Ronnie Toddywala, Nostrum Technologies: The device approval process is continuing to evolve, though the basic principles behind the process have not changed. The 510(k) has undergone a number of changes, to keep abreast with changing circumstances, and to cover some of the evolving medical landscape. There has been a significant increase in clinical testing requirements, and this is increasing the cost and complexity of drug development.
How can emerging device companies stay on top of regulatory trends and requirements? By performing internal audits on a periodic basis, scanning regulatory environment for changes and updates that can affect us, putting in significant effort on training our employees, and effectively managing our partners and suppliers. Another advice would be to implement technology solutions which can help us track, address and in the future prevent issues.
Complaint handling is a huge challenge. There are a lot of warning letters written for CAPA. Companies need to get smart about their complaint handling software, make sure it’s cloud-based, and that it can help determine regulatory reporting from each geography, have features such as complaint trending etc.

Impact of medical devices user fees
Trunzo: In 2012, the medical device industry user fee program was reauthorized for the third time. The program has set performance metrics in return for resources allocation from FDA. The program has planned for 200 additional review staff over the 5-year period, and now, at the end of the third year, most of these reviewers should have been hired.
For the industry, FDA committed to certain performance metrics to ensure greater interaction with the sponsor throughout the review process. To ensure that the interaction occurred, one of the metrics requires the FDA to have a substantive interaction goal between the sponsor and the agency. For instance, if it’s an application for a 510(k) approval, the company should receive some sort of communication from the reviewer by Day 60 – this could be either a request for additional info, or communication saying everything is on track for the review to be completed within 90 days. For PMAs, the deadline would be 90 days.
This is also the first time in user fee program that there has been a goal introduced that that measures the total time – from acceptance of application to time the decision is made. Our goal is to measure that average total time to the decision for both 510ks and PMAs. For 510ks, we are seeing modest improvement in average review times. For PMAs, because there is such a big lag time, those FY 13 and 14 cohorts do not yet have enough decisions to make an assessment to see if average time for PMAs is improving.
Which technology trend had the biggest impact this year?
Toddywala: I see a continued use of smart-phone based technologies to enable healthcare. Such technology will continue to evolve from being prevention and health focused – for instance monitoring steps to one takes – to becoming those that can modify and monitor changes in health behavior. I also see technology being used to enable patient compliance – for instance, SmartCap in medicine bottles which let loved ones or doctors know if someone is taking their medication as they are supposed to. Another area which will see growth is technology enablers for growing elderly population.
Nagarajan: The biggest trend I am noticing in working with a number of device customers is increasing reimbursement pressure, which has in turn, raised a focus on the economic value of the device or intervention, and on driving operational efficiencies. Another big trend is competition increasingly coming from non-traditional healthcare players such as Apple and Google. I often notice medical device companies organizing training programs focused on “If Google were to come after you, how would you respond,” to prepare themselves. 3D printing is another area that’s growing and will continue to have great benefits.
Trunzo: From a regulatory perspective, I believe, we are going to see more action from FDA because of focus on post market data collection. The agency is also establishing public-private partnerships with academic institutions to establish registries for product information and see how these can link with international registries. Also we can expect more guidance from FDA on healthcare tracking apps and technologies this year.
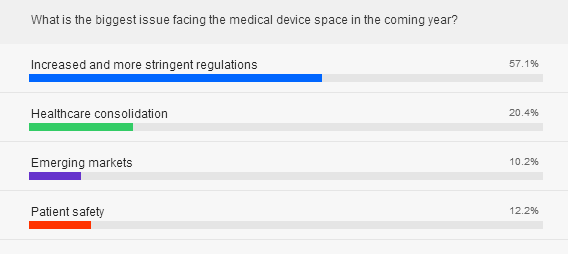
What should the industry be prepared going forward?
Nagarajan: As healthcare providers and care givers are increasingly resorting remote monitoring as a way to reduce healthcare costs, there will be an increased focus on data security and scrutiny. We can get greater agency scrutiny on how well we monitor all this information and keep them secure.
Toddywala: There is a continued evolution of requirements from FDA and other regulatory authorities and I expect this to increase in 2015 as the industry continues to develop more complex products.
Trunzo: Increased communication among global regulators will be an issue to contend with. Regulators across the globe talk to each other, and now many emerging markets are beginning to establish their own regulatory systems. The challenge for all of us will be to see more regulatory harmonization among all these regulators to ensure there isn’t much duplication.
For more on this discussion, click here.
Related Articles
-

In response to safety concerns and proposed regulatory action related to industrial use of ethylene oxide (EtO), the FDA Center for Devices and Radiological Health is taking additional steps to advance innovation in medical device sterilization, including recognition of new…
-

The UK MHRA has published “Software and Artificial Intelligence as a Medical Device.” The guidance document assembles previous guidances and regulatory requirements for SaMD and AIaMD devices seeking commercialization in the UK market.
-

Cybersecurity in health care is anything but simple. But significant changes can be expected in the coming years.
-

“Addressing unmet needs across pediatric populations is critical to advancing children’s health, and we are delighted to once again work with pioneering companies that seek to bridge this care gap."