REACH, which stands for the Regulation, Evaluation, Authorization and Restriction of Chemicals, is a standard that was established by the European Union (EU) in 2007 but does not go into effect until 2018. Simply put, REACH immediately seeks to “limit or prohibit the use of toxic substances in products.” According to an article written by Russell McCann (cofounder and CEO of Actio Corporation) in September of 2010, a time in which the REACH Regulation was “still rising to its crest,” the goals of REACH are divided into two categories: immediate and long-term.1 The long-term goal of REACH is that every person/body that needs to utilize chemicals will have access to the information that allows for safe usage. The standard aims to improve and clarify the legal aspects of chemical-related manufacturing and distribution processes occurring across the European Union.1
Naturally, legislation that deals with chemical usage will have an effect on the production of medical devices. Specifically, REACH regulation requires “producers of medical devices, or their chemical suppliers, to examine and disclose the characteristics of the substances they use, permanently defend the continued use of substances that are particularly dangerous, and in some cases, even face an outright ban on them.”2 The requirements imposed on medical devices by REACH differ depending on both the type of device and the nature of their arrival in the EU (i.e., whether or not they were manufactured in the EU).
According to REACH regulation, two categories of devices exist: preparations and articles. The classification of a device depends on the degree to which its chemical composition determines its function. Preparations are defined as “mixtures or solutions of two or more substances,” such as lubricants, bone cements, and anti-clotting agents; articles are defined as objects with “a special shape, surface or design” that primarily determines their function, such as cardiac pacemakers, catheters, corrective glasses, surgical gloves and defibrillators. The extent to which REACH Regulation applies to medical devices depends on their classification.2
Manufacturers of articles and both manufacturers and importers of preparations must disclose any information regarding the hazardousness of their devices. Furthermore, certain chemicals on the European Chemical Agency’s Candidate List of substances of very high concern for Authorisation may require prior authorization. Manufacturers, importers, and suppliers that utilize more than 0.1% of these substances must disclose this information to the health institutions as well as consumers that utilize their devices. Lastly, substances that are manufactured or imported in annual amounts of one ton or more must be registered, which requires submission of a technical dossier regarding toxicity. If the substance is manufactured in annual amounts of ten tons or more, registration also requires “a chemical safety report assessing the hazards, exposure, and risks of use during the [substance’s] entire life cycle.”2
Due to the nature of medical device composition, REACH Regulation, which specifically aims to address chemical usage, has greatly affected the manufacturing, importing, and distribution of medical devices across the European Union. REACH has also helped clarify the framework for where the responsibility lies when chemical issues result in complications within the medical device field. Among the affected products in wide distribution in the EU are disposable medical devices and durable medical equipment. Disposable medical devices include devices that are designed for one-time single-use on a patient; thereafter, it is either disposed of or recycled. Some examples of disposable medical devices are surgical drills, biopsy forceps, electrophysiology catheters and laparoscopy scissors. Durable medical equipment is medical equipment that is used in the home setting to improve quality of life. Examples include oxygen tents, nebulizers, blood sugar monitors, oxygen equipment and accessories. The remainder of this article addresses the applications of the REACH regulation for both disposable devices and durable medical equipment, although the steps to compliance are common to most medical devices. Compliance steps include substance identification, registration, evaluation, authorization and restriction.
Substance Identification
Substance identification is the prerequisite for the majority of REACH processes. The benefits of the correct identification of substances include:
- Preventing unnecessary replication of the testing on animals
- Cost effectiveness
- Providing information on composition and purity of substances
- Ensuring clear and concise description of analytical methods
The legal reference for substance identity is shown in Annex VI item 2 of REACH.3 Useful information about how to name and identify chemical substances based on REACH can be found in ‘Guidance for identification and naming of substances under REACH and CLP on the European Chemicals Agency website.
Registration
Companies are responsible for assessing hazards and potential risks, and for collecting the properties and usage information of the substances they manufacture or import, at or above one ton per year. The collected information is communicated to the European Chemicals Agency (ECHA) through a registration dossier containing hazard information. The assessment may lead to a result indicating how these risks should be controlled.
Registration for medical devices requires manufacturers or importers to submit key items for consideration, including a technical dossier with data on toxicity and eco-toxic properties of the substance. A chemical safety report with assessment of hazards, exposure, and risks of use during the entire life cycle of the substance in quantities of 10 tons or more per year is also required.
Registration follows the “one substance, one registration principle”. Manufacturers and importers of the same substance should submit their registration jointly. The substance identity should be consistent and sufficient information should be provided to confirm the substance identity. There is typically a substance registration fee surcharge.
Evaluation
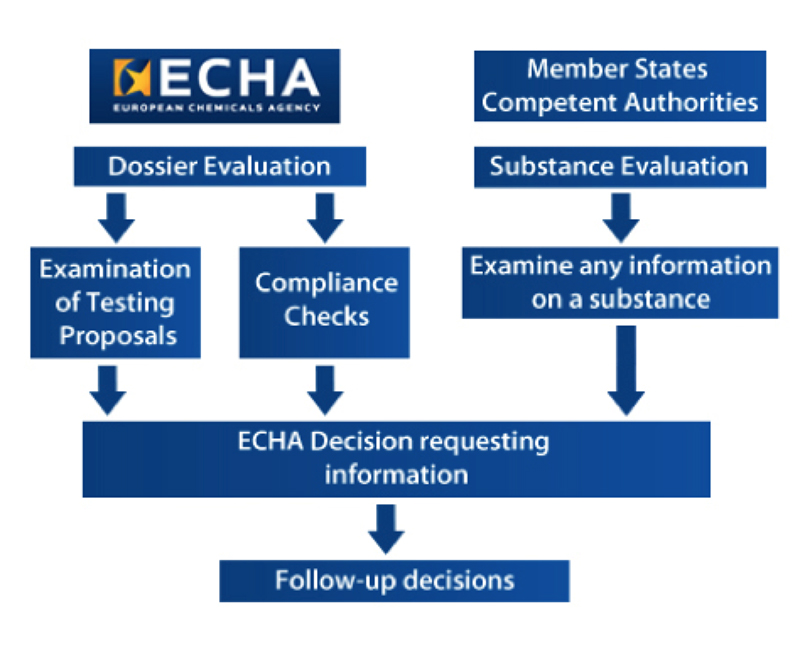
The European Chemicals Agency provides the overview of the process of the Evaluation (see Figure 1).
The three areas of evaluation are:
- Examine the testing proposals submitted by the registrants
- Check the dossier submitted by registrants
- Substance evaluation
Subsequent to the initial evaluation, there may be further requirements for the registrants to submit more information on the substance.
REACH Resources
REACH and Its Impact on Medical Devices. (2010). Covington & Burling LLP. September 2008
European Parliament Press Release REACH – MEPs debate chemicals legislation and new Chemicals Agency
European Chemicals Agency (ECHA)
Guidance for Identification and Naming of Substances Under Reach and CLP
Authorization
The authorization procedure is used to regulate the risks form, Substances of Very High Concern (SVHCs), and ensure that the SVHCs are properly controlled. Under the condition of proper function of the EU internal market, the SVHCs will be progressively replaced by appropriate alternatives.
To be identified as SVHCs, a substance should meet the criteria for classification as carcinogenic, mutagenic or toxic for reproduction category 1A or 1B. The substance should be persistent, bioaccumulative and toxic (PBT) or very persistent and very bioaccumulative (vPvB) based on Annex XIII of the REACH standard. SVHCs may be included in the Authorisation List after the two-step regulatory process; manufactures, importers, and downstream users can apply for authorisation if they are on the Authorisation List.
The EU manufacturer of medical devices must ensure that they apply for authorization of the listed substances in specific concentrations, (e.g., 0.1 % in the manufacturing process of the medical device). Imported SVHCs in medical devices with article forms such as equipment are not subject to authorization requirements.
Applicants who do not obtain an authorization will be banned from using the listed substance unless their downstream users or suppliers have already obtained the same authorization. For each authorized medical device or substance obtained by manufacturers and importers, a number of authorizations will be given and labeled.
Restriction
Restriction is used to avoid the unacceptable risks posed by chemicals. Restrictions may limit or ban the manufacture, import and placing on the market of a particular substance or substances. A member state, or ECHA, can propose restrictions if the risks need to be addressed on a union-wide basis. ECHA also can propose restrictions on substances noted on the Authorisation List mentioned in Annex XIV.
When proposing a restriction, ECHA will work together with the appropriate Member State(s) to provide scientific opinions that will help the European Commission to make robust final decisions.
While the new regulations have certainly made medical device production and distribution standards more stringent in the EU, REACH has created a more regulated and thus, safer device industry. More detailed information can be found on the European Chemical Agency official website.
References
- McCann, Russell. “REACH Regulation: The 5 Most Commonly Asked Questions.” Environmental Leader. N.p., 2 Sept. 2010. Web. 27 April 2015.
- Bogaert, Peter, Richard Kingham, and Candido Garcia Molyneux. “REACH And Its Impact on Medical Devices.” Covington and Burlington LLP (2008): 1-5. Sept. 2008. Web. 27 Apr. 2015.
- Regulation of The European Parliament and of The Council on Classification, Labelling and Packaging of Substances and Mixtures, Amending and Repealing Directives 67/548/EEC and 1999/45/EC, and Amending Regulation (EC) No 1907/2006, Annex Vi: Harmonised Classification and Labelling for Certain Hazardous Substances.
Related Articles
-

The draft guidance proposes updates to clarify how the Breakthrough Devices Program may be applicable…
-

The updated guidance clarifies how the program applies to medical devices that may address health…
-

With the ever-increasing adoption of connected devices, the agency is emphasizing the need for effective…
-

In addition to challenges brought by new EU MDR, Switzerland has struggled to approve and…