

As device organizations continue to evolve in the integration of combination products CGMP’s into their business, a holistic approach can prevent many unnecessary delays. This article provides some insights to consider when integrating stability testing into your device Quality System Requirements (QSR) system. It will discuss a number of quality systems that Stability may significantly impact. Part of the holistic approach should be a discussion that looks out to five years from today and defines where does the organization wants to be with regards to the management, development, manufacturing and compliance of their combination products business. The following six combination product requirements should have good quantitative measures for this five-year plan.
- Delivery
- Packaging/Labeling codes and the expected order fill rate. If you have five codes that are expected to be delivered to customers on a monthly basis, the delivery challenge will be different than coordinating the manufacture of a 100 codes that will be delivered weekly. The impact of drug release testing and approvals can significantly impact your delivery objectives. Drug testing failure investigations may take longer than expected.
- Safety & Efficacy
- The drug and combination product complaint and serious complaint (SAE/MDR) profile should be understood. This information should be incorporated into the risk management models.
- Identity
- This must be addressed for every lot. Make sure the drug definitions of lot and batch are well understood, this may affect the number of tests to be conducted and the infrastructure required.
- Strength
- How the device affects the strength of the drug may require special analytical methods. These analytical methods should meet ICH Q2 Analytical Methods.
- Purity
- Identifying all the impurities and degradants for both device and drug will be important. Also any process that can affect the impurities and/or degradants needs to be well understood and a control strategy implemented.
- Quality
- Once the drug and the device are combined the combination product quality characteristics must be defined. There can be effects due to storage, environment, contact time between the device and the drug and use.. A useful exercise is to match the critical-to-quality (CQA) characteristics with relevant product complaints. A control strategy that addresses the CQA’s of both product and process characteristics could be a good source of preventative measures.
It is important to identify key functions affected by these requirements (i.e. Stability) and assess the functions ability to meet them consistently. Part of the strategy should ensure that these functions have the ability to consistently meet the combination product requirements.
Figure 1 is a framework that can be used to help organizations do the following:
- Think about the affected functions, systems and infrastructures needed to manufacture the combination product
- Develop a plan to prioritize what will be done and when
- Include in the plan criteria for sustainable solutions to be implemented and monitored
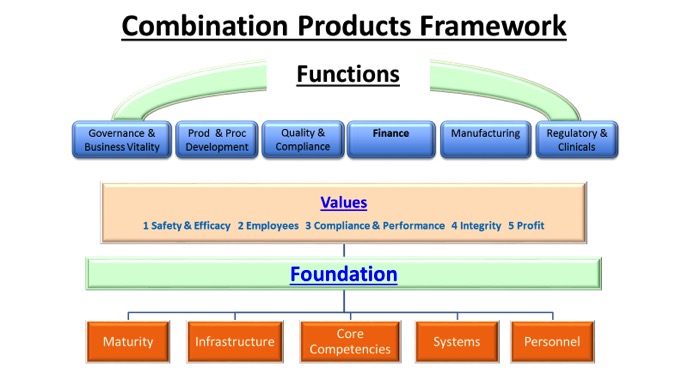
Integrating a new set of CGMP’s (21CFR 820) into an organization that is already operating to a well-established set of QSR’s (21 CFR 820) can be disruptive. Often some basic planning and analysis can make a significant difference in the implementation effectiveness. Before beginning to evaluate the organization’s ability to integrate these new combination product 211 CGMPs into your quality system determine the foundation that would be necessary for each CGMP (21 CFR Part 4) you plan to implement. Once you decide what are the systems, personnel and infrastructure needed, then ensure your organizations relevant core competencies can address these needs. Also assess the maturity and consistency of their performance or at what stage of experience they are operating.
Exclusive VIDEO: How to Gather Clinical Evidence for Combination Products | WATCH NOWA good understanding of how the CGMP’s defined in 21 CFR Part 4 can impact the values of the organization can help you proactively address personnel concerns. Device manufacturers have a good understanding of the safety and efficacy of their products; however, these new CGMP’s may lead you to ask questions you did not previously consider (i.e., questions about the control of degradants, stability indicating methods, reserve samples, control of pyrogen). Employees may see these new requirements as a threat to the effective execution of their functional responsibilities. You may find that maintaining your compliance performance is impacted and some support is needed to get compliance to the level at which you are accustomed to operating.
The expectation is that these new CGMP’s will long term impact profits; temporarily there may be an impact. This should be defined and discussed early in the process.
In particular you may need to change or add facilities and/or new equipment. As you begin to pull together all of these CP CGMP insights that may impact your values, foundation and functions together, you must be specific what is expected of each function. An important aspect of your plan will be to make sure key stakeholders in impacted functions are on board with the approach. Make sure they agree with the need, the plan, what is acceptable and when this level of performance will be in place.
Many device organizations have chosen the Streamline approach for their compliance with the combination products CGMPs. Specifically, the streamlined approach provides that a device organization manufacturing combination products may meet the requirements of both the drug CGMPs and device QS regulation by designing and implementing a CGMP operating system that is demonstrated to comply with the following:
(i) 21 CFR 211.84. Testing and approval or rejection of components, drug product containers, and closures (ii) 21 CFR 211.103. Calculation of yield (iii) 21 CFR 211.132. Tamper-evident packaging requirements for over-the- counter (OTC) human drug products (iv) 21 CFR 211.137. Expiration dating (v) 21 CFR 211.165. Testing and release for distribution (vi) 21 CFR 211.166. Stability testing (vii) 21 CFR 211.167. Special testing requirements (viii) 21 CFR 211.170. Reserve samples |
A requirement for 211.137 Expiration dating is to have it determined by appropriate stability testing. For single-entity combination products, you will need to define the applicable standard of drug identity, strength, quality and purity. Most device organizations are well prepared for the quality and identity requirements; however, strength and purity can be difficult. How you measure the strength of the drug when it is physically or chemically combined with a device and how you define the specification can be challenging. Another area that has presented some degree of difficulty is purity, particularly understanding the degradant products of the drug and how the processing affects the degradants of the combination product.
A helpful FDA technical guidance the device community should understand is “INSPECTIONAL TECHNICAL GUIDANCE; Expiration Dating and Stability Testing for Human Drug Products, 10/18/85 Number: 41. The agency provides guidance on key areas of Expiration Dating and Stability Testing. Some of the areas they cover are:
EXPIRATION DATING
- Absence of an Expiration Date will result in regulatory action
- OTC Drug Exemptions
- Products Intended for Reconstitution ED and Stability requirements.
STABILITY TESTING
- Impact of no Written Stability Testing Program
- Supportive Stability Data
- Number and Size of Batches
- Three Batch rule and changes
- Number and Size of Batches
- Accelerated Studies
- Appropriate use of accelerated studies are discussed and certain uses of accelerated studies that are discouraged
- Test Intervals
- Normal interval – Initial, 3,6,9,12,18,24,36
- Annual Testing
- Storage Conditions
- Room Temp defined as 24-250 C. Products liable to degradation by light or moisture, testing at these conditions needs to be evaluated and addressed.
- Test Methods
- Although 211,194 is not called out, review its applicability and confirm your record system meets this requirement, particularly Section 211.194 (a) (2) verification under actual conditions of use.. Also will need to have in place stability indicating methods. This will require a good understanding of the degradants in your combination product.
- Container-Closure Systems
- When changing the package or unit the drug is stored in, ways to do satisfactory comparison of container-closure systems
- Container Sizes to be Tested
- For more guidance also see ICH Q1D Bracketing and Matrixing Designs for Stability Testing of New Drug Substances and Products Preservatives
- Preservatives
- Guidance for products formulated to contain preservatives to inhibit microbial growth
- Bulk Drug Substances (Bulk Pharmaceutical Chemicals)
- Guidance for stability testing program for bulk drug substances
- Sterility Testing
- Guidance on container-closure system that has demonstrated ability to maintain sterility throughout the expiration dating period and revalidation when other ingredients are added
To help you design a stability program you should consult experts on how to incorporate the applicable sections of the ICH Quality guidances into your program:
- Start with a commitment to gain a working knowledge of all applicable USP General Notices, Monographs & Chapters.
- At a minimum, perform a thorough review of the following ICH guidances and identify what sections need to be addressed in your stability, expiry dating and reserve sample practices. Sections that do apply but have not been addressed should be discussed with experts in this area:
- Q1A – Q1F Stability
- Q2 Analytical Validation
- Q3A – Q3D Impurities
- Q4 – Q4B Pharmacopoeias
- Q6A- Q6B Specifications
- As you incorporate these guidances into your organization, an in-depth review of your laboratory practices and the labs’ ability to meet the requirements of 211.194 would also be helpful, particularly if your labs are testing drug products.
- Review FDA’s “Investigating Out-of-Specification (OOS) Test Results for Pharmaceutical Production” Guidance and confirm your laboratory nonconformance practices would be able to meet the practices identified in this guidance.
- Pay attention to how you manage stability trends, and evaluate historical data for the batch on stability as well as the overall product. When you find an out-of-trend indication, an investigation must take place.
- If your combination product requires an NDA/ANDA, and you choose to keep your QSR system as the base Quality System, one of the many CGMP systems you will need to understand is the requirements of Annual Product Review.
- Note for stability testing that the product must be in its final packaging. If this is not the case the FDA should be consulted. There are instances in which the agency has accepted other practices.
- Have a good rationale for which product codes are included in the annual stability testing, which is particularly important when there are a large number of product codes.
Related Articles
-

The creation of the Office of Combination Products more than a decade ago may have…
-

Susan Alpert, principal of SFA Consulting, explains the role of clinical evidence for combination products…
-

The agency is creating a virtual center of excellence to expedite the development of combination…
-
Combination products are a major growth area for life science companies and in the future,…
-

With the goal of acting as a single agency, FDA’s OCP has several activities planned…
