

This series of articles explains how an all-encompassing testing approach will improve product reliability and secure your bottom line.
The medical device manufacturing industry is subject to regulations to ensure their sterile packaging maintains the integrity of that sterile barrier throughout the supply chain. Furthermore, the packaging must protect the product throughout the distribution cycle. The evaluation of product and package integrity following simulated distribution has inputs from Process Validation, Risk Management, Design Validation, and Test Method Validation, but is at its purest, a functional requirement of Design Verification. This article presents a compliant and operationally efficient approach to design verification of product packaging throughout the supply chain, all the way from the manufacturing floor to the eventual use by a customer. This series of articles will argue that this fully encompassing testing improves the reliability of the medical device and ultimately secures the manufacturer’s bottom line. The costs of this initiative are more than offset by the avoidance of regulatory action, product liability and long-term improvements to the manufacturer’s image. First, this series will describe how a device manufacturer could conduct design verification supply chain activities, by considering elements of material construction, process capability, and test method precision/accuracy. Second, it will argue that this approach benefits the manufacturer by ensuring compliance with regulations, increasing efficiency through industry best practices, and reducing the impact of negative post-market outcomes on the patient population over the long term.
Design Considerations Prior to Testing
The purpose of this type of design verification is to evaluate the effectiveness of the packaging configuration at maintaining the sterility of the product within and protecting the product from damage throughout the distribution cycle. The most common mistake when undertaking this testing is that often, manufacturers stop short of combining the conditions of the entire supply chain on the packaged product. This can lead to distribution-related product failures and even non-sterility of the product, forcing field actions and regulatory consequences, in addition to risking patient safety. When approaching the design verification study, there are several elements to consider in order to ensure a complete assessment throughout the supply chain.
1: Where will you ship the product?
Some regions, such as Japan, have particular regulatory requirements for product aging, which can challenge the package integrity and must be included in the test regime in order to be accepted for sale in the Pacific Rim region. Other global locations have particularly challenging terrain or temperature extremes that must be considered. Before beginning to write the test protocol, it is important to establish the distribution goals for the product family, both now and into the near future, as it is more cost effective to include those requirements in one series of testing than to repeat the entire test sequence later. The answer to this question will determine which simulated distribution standard should be used. For example, if shipping an individually packaged product of 150 pounds or less internationally, the ISTA 2A standard is one possible choice, but the ISTA 1A standard is not appropriate for international shipping distribution simulation testing. Many manufacturers have made this error and some notified bodies have forced them to repeat design verification testing on their packaged products, using the correct standard, in order to retain their CE Marks. Consider the shipping methods that may be required to access those global regions. For example, will there be air travel involved, or travel by sea? Ocean travel brings high humidity, air travel can involve temperature and pressure extremes, travel by rail can induce harmonic frequencies that may exacerbate package and product damage, and travel by truck is subject to road conditions. This will determine, within the selected standard, which test sequence to perform. For example, let us say you are using the ASTM D4169 standard, and your product is subject to shipping by truck and rail. Distribution Cycle 13 (ASTM D4169-05, DC=13) includes drop, compression, vehicle stacking, and vibration simulations in its test sequence, which would be suitable for packaged products being delivered in trucks and trains.
ASTM D 4169 “Standard Practice for Performance Testing of Shipping Containers and Systems” is performed by subjecting shipping units to a test plan consisting of a sequence of hazard elements that may be encountered in various distribution environments. A simple example would be shock testing followed by drop testing, then vibration and finally compression testing. The test plan provides a uniform basis of evaluating in a controlled and repeatable laboratory environment the ability of the shipping units and contents to withstand the distribution environment. The test plan uses established testing services methods at levels representative of those encountered in actual distribution. The Distribution Cycle (DC) most commonly used for medical device packages is DC-13, which is designed for the small parcel and overnight shipping mode. Design customized distribution cycles when the anticipated distribution of the product is well understood and defined.
ISTA Pre-Shipment Test Procedure for Packages utilizes tests simulating the shocks and stresses normally encountered during handling and transportation; these tests provide a means for a manufacturer to assign the probability of safe arrival for their packaged products.1
2: Risk Management
Risk management, as currently prescribed by ISO 14971:2012, lays out several requirements for medical devices. In particular, the hazard analysis, including potential failure modes and hazards under both normal and fault conditions, and the risk analysis or failure mode and effects analysis (FMEA), are pertinent to the design verification study. Further, a review of post-market performance by similarly packaged product can provide insight into the potential failure modes and design considerations prior to freezing the package design. Use the design FMEA or its equivalent to define risk-based acceptance criteria for the study protocol. For example, loss of product sterility typically has a high-risk profile (severity rating). Therefore, we assign a reliability of 99% and a confidence level of 95% to acceptance criteria for sterility, whereas we may assign a cosmetic defect, such as a slightly crushed external shipper corner, a reliability of 80% and a confidence level of 20%.
3: Sample Size
In preparing to generate the test protocol for the design verification, the sample size is one criterion that seems to cause undue stress. If the design FMEA is up to date, and there is a corporate procedure aligning risk levels with quantitative acceptance criteria such as reliability and confidence level, select a sampling plan that aligns with the specified reliability and confidence level. These sampling plans are published widely in the public domain and can be derived from the Operating Characteristic (OC) curve, wherein the reliability of a given sampling plan is associated with its Lot Tolerance Percent Defective (LTPD) value for a given confidence level. Many companies consider distribution simulation testing to be a challenge condition, and therefore they justify the use of lower reliability and smaller sample sizes. Be certain that the test protocol includes a statement about challenge conditions, if applicable, and cites the risk level, reliability, confidence level and sample size for each test condition. Indicate whether you will use variable or attribute sampling plans.
4: Bookending based on product and package design
Document the unit rationale in the test protocol. At times, we can consider a single design to be representative of an entire product family based on the size and number of sterile pouches included in each carton. Another approach is to select the smallest and largest, or lightest and heaviest products to “bookend” the range of interactions between product and package for the study.
5: Actual or dummy units
If the product will undergo functional testing following simulated distribution and aging, it should be produced under normal production conditions with production-level components and operators and methods, or equivalent. This article gives a method for verifying the product-package performance throughout the entire supply chain and shelf life, which requires multiple test groups. It may be advantageous to include functional product in some of the groups (i.e., wherever functional product testing will be performed following simulated distribution or aging studies), but not necessarily all groups. For example, let us say that in order to release your product commercially, you need a minimum shelf life of one year, but you would prefer to verify a two-year shelf life if possible. In this case, you would need to include functional product for both groups as insurance. For test groups not requiring functional product testing, say for verification of a change to packaging materials where product functional testing was previously verified, substitute dummy product to simulate the profile and weight of the packaged product. These might be rejected units with all attached components, for example.
6: Ensure product and package material tolerance
Ensure the tolerance of product and package materials to sterilization method, aging conditions and the high temperature of accelerating aging in ovens. Consider a full characterization of materials of construction of the product and package, along with the required shelf life and sterilization method. Often the external parameters define the suite of materials available for product design as much as the device’s inherent performance requirements. There is an example of a thoughtfully designed, packaged product that was compatible with the changes in temperature, pressure and humidity associated with the ethylene oxide sterilization cycle, and was stable for the shelf life of the product. However, when the engineer selected the oven temperature for accelerated aging, he failed to realize the temperature exceeded the glass transition temperature of the main polymer of construction, rendering the devices untestable. Do not select a 60°C AA temperature if your product or packaging materials can only withstand 40°C!
7: Shelf life
As noted above, shelf-life testing is a requirement of the regulations prior to releasing medical devices for commercial distribution. The widespread use of accelerated aging demonstrates the stability of the materials of construction of both the packaging and the product over time. Accelerated aging is based on the assumption that chemical reactions in materials follow the Arrhenius reaction rate function. Based on modeling kinetics of materials, this function states that every 10°C increase or decrease in temperature of a homogenous process results in a 2x or 1/2x change, respectively, in the rate of a chemical reaction. This chemical reaction rate is known as Q10, and is denoted Q10=2. This applies to shelf life as the equation:
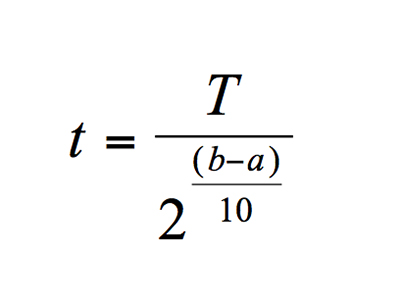
The next article in the series describes process considerations prior to design verification of packaged products.
Reference
- Distribution Dynamics Certified Testing Laboratory. “Package Testing, Product Testing and Materials Testing for the Medical Device Industry”. Retrieved from: http://www.testedandproven.com/
Related Articles
-

The acquisition expands Veranex’s footprint for product design engineering services across the U.S., with combined…
-

Hägen, who was a contributor to Medtech Intelligence, spent 28 years building BlackHägen Design to…
-

The acquisition expands Veranex’s European footprint as well as the scale and scope of its…
-

Bringing design and quality assurance processes together earlier in the device development process can reduce…