

While companies currently must demonstrate a post-market surveillance plan for CE-marking, the European Union Medical Devices Regulation (2017/745) increases requirements for post-market surveillance (PMS). Articles 82 through 86 and Annex III of the EU MDR outline the requirements for a post-market surveillance system (with other requirements peppered throughout the regulation) resulting in numerous changes to your existing practices. Gone are the days when a manufacturer could point to their passive complaint and feedback system to address PMS requirements—although you may have already noted this to be the case in your MDSAP or ISO 13485:2016 audits. This article presents an evaluation of these changes and presents a hierarchical outline of recommended actions to comply.
Procedure
Article 83 requires an active post-market surveillance system for each device that is “an integral part of the manufacturer’s system”. This means that your post-market surveillance (PMS) system must be adequately documented in your quality management system (QMS) and that PMS activities are effectively linked to other areas of the QMS. When drafting or revising your PMS procedure, ensure appropriate linkages with the following:
- Design and Development (where post-market surveillance information may be relevant for products in development)
- Feedback and Complaint Handling
- Field Action (Recalls and Safety Notices)
- Management Review
- Risk Management
- Technical Documentation
- Labeling
- Clinical Evaluation
- Corrective and Preventive Action
- Periodic Safety Update Report (if applicable, a new requirement of the EU MDR)
- Summary of Safety and Clinical Performance (if applicable, another new requirement of the EU MDR)
- Regulatory Communication (in addressing compliance to ISO 13485:2016, you may have already strengthened your procedures for communicating with regulatory agencies).
Any QMS procedure whereby product performance data is gathered or evaluated must be linked to your post-market surveillance procedure.
All of these new requirements and interacting systems may seem like a daunting challenge. To begin this process, map out your existing processes and then evaluate where changes are needed. Consider using this activity to also develop a new QMS process interaction diagram. If you don’t already maintain such a diagram, this is a graphical depiction (e.g. flowchart) of the QMS processes and their interaction. By documenting this diagram as an appendix to your quality manual, you provide high-level evidence of a process approach as defined in ISO 13485:2016 and will succinctly depict the process interactions required by the EU MDR. While the flowchart below is simply an example, perhaps you will derive some ideas from the following example relating to your PMS system and linked processes.
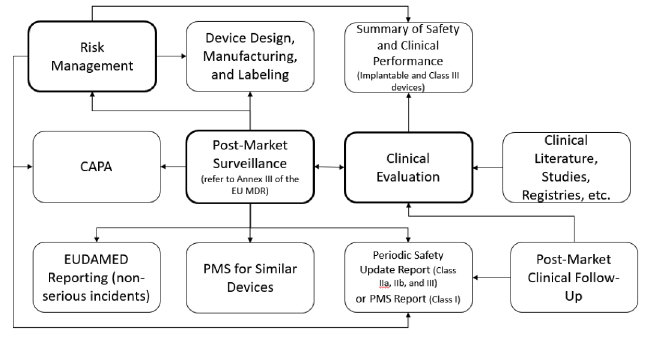
In the above diagram, the processes I consider “primary” are in bold. By “primary”, I mean these processes are complicated or significant enough to warrant their own procedure. For these procedures, consider how you might:
- Develop a new post-market surveillance procedure that includes all requirements from Articles 82-86 and Annex III
- Revise your existing Clinical Evaluation and Risk Management procedures to address new requirements (particularly Post-Market Clinical Follow-Up and Summary of Safety and Clinical Performance)
- Coordinate the activities performed under each procedure. With the exception of CAPA, you may want to document other processes (non-bold) in the procedures for the “primary” processes (bold) for simplicity and efficiency. For example:
- Define requirements and the process for PMCF in your clinical evaluation procedure
- Identify reporting requirements (PSUR and PMS Report) in the PMS procedure
- Move existing complaint trending practices into your PMS procedure
A “swim lane” approach is another way to organize existing and new requirements. For example:
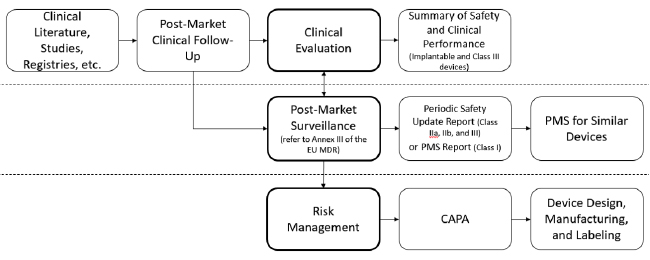
Again, there are many ways to address PMS and related processes—these are simply ideas for your consideration. Regardless, mapping these processes is essential for effective procedures that address EU MDR requirements. With the added complication of the EU MDR, such graphical depictions are essential for planning, training and execution.
Because the EU MDR requires that post-market surveillance is proportionate to the risk class of the device, you’ll want to ensure that your procedure covers varying approaches that allow more rigorous post-market surveillance for higher-class devices. If you only manufacture one product or one class of product, your procedure need not address other device classes. However, you must document how the activities you perform are appropriate for the risk class of the device.
Plan
The procedure you create for PMS will be used to create your PMS Plan. The Plan is used to conduct the PMS process for each product. There must be post-market surveillance plan for each device and the plan must be an integral part of your QMS. This means that your post-market surveillance for each device must be planned, maintained, documented and updated in your QMS. The procedure linkages outlined in your procedure must be followed through with the requirement for data linkages in your plan.
The regulation does not provide guidance or details on the type of analysis or when data requires some type of action (corrective/preventive action, field action, etc.). It is up to you as the manufacturer to define the “triggers” that require evaluation for such action. Regardless, your post-market data must be used to update your risk management. Since your risk management system already identifies the rate and severity of potential device problems, consider how you might address these two requirements with one risk process.
Since post-market and regulatory reporting must be “carried out within the manufacturer’s organisation by a person responsible for regulatory compliance who fulfils minimum conditions of qualification”, the planning stage is the latest point at which your organization should bring this expert (or experts) into the process. A contractor or organization may provide you with a complaint procedure, but the planning and execution of the post-market activities must be managed by your resident regulatory compliance experts. Consider this resource requirement when planning for the transition!
Articles 83 and 84 briefly describe the need for a PMS plan. This plan must be included in your technical documentation (and will therefore be assessed as part of your technical file/design dossier review for CE-marking). The detailed requirements for the plan are laid out in Annex III of the EU MDR. Since Annex III is a straightforward listing of requirements, I won’t repeat them here; however, I recommend pausing here and referring to Annex III prior to continuing with the recommendations below.
Process
The PMS process is the collection and analysis of the data collected from the various sources outlined in Annex III and conducted according to your PMS plan. This process of collecting and analyzing data must be active, systematic, and throughout the device lifetime. Do not confuse device lifetime in this context with service life or shelf life. Data collected as part of the PMS must be defined in the plan and used for the following:
- Evaluating and monitoring for corrective and preventive action. Consider how best to link PMS to CAPA—a passive linkage (meaning a CAPA is initiated only when an obvious breakdown is realized through PMS) will not suffice. When evaluating the results of your PMS program, you should look for nonconformances and potential nonconformances. For any nonconformances or potential product problems, ensure you maintain a documented evaluation, including any conclusions drawn regarding the need to conduct field action (recall).
- Conducting regulatory reporting. A statistically significant increase in the frequency of non-serious events (serious incidents already require reporting to Competent Authorities via the EU Vigilance system) will need to need to be reported via EUDAMED. As you may be aware, EUDAMED as described in the EU MDR has not yet been implemented. However, you should nonetheless address this requirement in your procedures.
- Improving the device and updating technical documentation. Consider also using your CAPA process to meet another requirement of the EU MDR—updating design, labeling and manufacturing information for the device in response to PMS data. The CAPA system may be used to document the need for changes to the design, labeling or manufacturing and the plan for doing so. Such changes include those that improve the safety, performance and usability of the device.
- Updating Risk Management. Data gathered in post-market surveillance must be used to update the risk/benefit profile of the device and improve risk management for the device. The most practical way to achieve this is to coordinate your periodic risk management review with your evaluation of post-market surveillance data. Verify that any new hazards or harms are incorporated into your risk evaluation and that any event rates are evaluated for corrective action.
- Evaluating for other relevant products. Ensure that PMS for each device is also evaluated in light of similar devices, other devices in the product family, or wherever the data may be relevant to other devices. As you evaluate PMS data, consider how you might flag data relevant to other products. You can review this flagged information when performing the PMS review for those products.
- Updating the Summary of Safety and Clinical Performance. For implantable and class III devices (excluding custom or investigational devices), you must maintain a Summary of Safety and Clinical Performance. Minimum requirements for the Summary of Safety and Clinical Performance are outlined in Article 32 of the EU MDR. All major clinical data processes (clinical evaluation, post-market clinical follow up, and post-market surveillance) feed into this report. Therefore, the data generated from these related activities will be used to update the summary of safety and clinical performance.
Remember that change enacted as a result of post-market surveillance may result in the “Butterfly effect”! Any changes must be evaluated for impact elsewhere and potential revalidation, updates to the technical documentation, or regulatory approval. However, your current change and document control practices should already address these requirements. Corrections and corrective actions must also be assessed for regulatory requirements (e.g., reporting of field actions conducted in response to PMS data).
Reporting
In addition to the clinical evaluation report, there are two new reports required by the MDR: the post-market surveillance report and the periodic safety update report (PSUR). These new reports are primarily intended for regulatory review and therefore must be “readily searchable” (maintained in a searchable electronic format).
For Class I devices, a post-market surveillance report is required to document the results of the post-market surveillance for each device. The data, analysis, and any conclusions are documented in this report. This report is updated “when necessary” (no specific timeline is given in the EU MDR) and is only provided to the competent authority upon request.
For Class IIa, IIb, and III devices, the results are documented in the periodic safety update report. PSURs for Class IIa devices must be updated at least every two years and PSURs for Class IIb and III devices must be updated annually at a minimum. For Class III and implantable devices, the report will be provided to the notified body for assessment and uploaded to EUDAMED (results of the notified body’s assessment will also be uploaded to EUDAMED).
Summary
Post-market requirements outlined in the EU MDR carry significant process challenges and procedure updates. Start simple and try not to overcomplicate your processes—the new requirements are complicated enough. Review the requirements laid out in Article 61 (Clinical Evaluation), Articles 83-86 (Post-Market Surveillance), Annex III (Post-Market Surveillance), and Annex XIV (Clinical Evaluation and Post-Market Clinical Follow-Up). Start with a process map of your existing systems and modify to meet new requirements. Assign compliance activities to the appropriate functional areas and let them determine how best to link processes. For example, assign your clinical research department procedure updates related to clinical evaluation and PMCF. Your dedicated regulatory compliance staff can manage the procedures for PMS process, planning and reporting (PMS Report and PSUR). Those functional areas can work together to best determine how to interface the clinical research responsibilities with the PMS requirements. Lastly, have an independent employee or consultant verify that your procedures meet all EU MDR requirements before use. By the end of this process, you’ll have a multi-disciplinary team of PMS experts ready to satisfy the regulatory requirements and serve as subject matter experts during regulatory audits.
Related Articles
-

The draft regulations, “The Medical Devices (Post-market Surveillance Requirements) (Amendment) (Great Britain) Regulations 2023,” serve as an amendment to the existing UK MDR Part 4A post market surveillance requirements, and are currently with the World trade Organisation (WTO) for member…
-

On Monday, March 20, the EU MDR extension approved on March 7 came into effect following its official publication in the Journal of the European Union.
-
The position paper from the European Commission’s Medical Device Coordination Group (MDCG) recommends an extended timeline to allow certain MDD or AIMDD certified legacy devices to come into compliance with MDR.
-

The two new final guidances are intended to increase transparency to stakeholders on the FDA’s approach to the issuance and tracking of 522 orders and post-approval study requirements.