

Production and operations managers—make sure your preventative maintenance procedures are up to snuff.
Production and operations managers—make sure your preventative maintenance procedures are up to snuff.
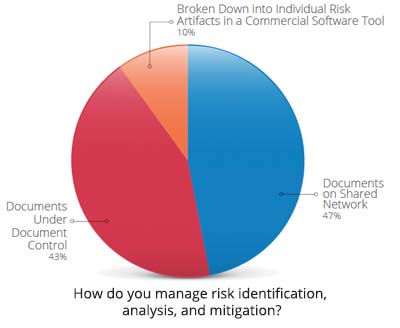
With the increased complexity of devices, a streamlined approach to managing product development risks and documenting compliance is challenging but perhaps more important than ever.
Some companies just don’t get it. If a device has been cleared or approved for a specific indication, it can only be advertised for that specific indication.
There are several steps you can take to protect your IP and avoid medical device piracy in China.

Differing approaches to risk could hamper further growth of combination products.

The creation of the Office of Combination Products more than a decade ago may have been a big step forward, but frustrations surrounding policy-making and coordination between CDRH, CDER and CBER remain.
Take a quick trip to the FDA’s warning letter page and you’ll see the potential danger that novelty lenses pose.

This year’s work plan includes several items that will impact medical device manufacturer and suppliers.

Roberta Goode, president & CEO of Goode Compliance International, weighs in on the latest challenges medical device manufacturers are facing in risk management at the MedTech Intelligence HHE, Risk Assessment, & Recalls Conference in Washington, DC.
REACH, which stands for the Regulation, Evaluation, Authorization and Restriction of Chemicals, is a standard that was established by the European Union (EU) in 2007 but does not go into effect until 2018. Simply put, REACH immediately seeks to “limit or prohibit the use of toxic substances in products.” According to an article written by…



